 可咨询
可咨询
擅长:缺铁性贫血、特发性血小板减少性紫癜、原发性血小板减少性紫瘢、免疫性血小板减少症、白血病、淋巴瘤、多发性骨髓瘤、骨髓增生异常综合征、慢性粒细胞性白血病、骨髓瘤
 可咨询
可咨询
擅长:贫血、再生障碍性贫血、缺铁性贫血、溶血性贫血、地中海贫血、营养性贫血、血液系统肿瘤、多发性骨髓瘤、骨髓增生异常综合征、白血病、淋巴瘤、骨髓瘤、慢性粒细胞性白血病、慢性白血病、急性淋巴细胞性白血病、骨髓增生性疾病、恶性淋巴瘤、紫癜、特发性血小板减少性紫癜、过敏性紫癜、原发性血小板减少性紫瘢、血小板减少性紫癜、血小板疾病、血小板减少症、血小板增多症、免疫性血小板减少症、凝血功能障碍、血友病、淋巴癌、白细胞减少症
 可咨询
可咨询
擅长:解读血常规异常、擅长疑难血液系统疾病的诊断、个性化给予良恶性血液病建议、贫血、血小板增多症、血小板减少症、白细胞增多、白细胞减少症、红细胞增多症、缺铁性贫血、溶血性贫血、地中海贫血、血小板减少性紫癜、营养性贫血、过敏性紫癜、特发性血小板减少性紫癜、免疫性血小板减少症、血液系统肿瘤、多发性骨髓瘤、淋巴瘤、骨髓增生异常综合征、急性淋巴细胞性白血病、恶性淋巴瘤、凝血功能障碍、急性白血病、急性淋巴细胞白血病、慢性淋巴细胞白血病、白血病前期、类白血病反应
 可咨询
可咨询
擅长:贫血、再生障碍性贫血、缺铁性贫血、溶血性贫血、营养性贫血、血小板疾病、血小板减少症、血液系统肿瘤、慢性白血病、骨髓瘤、白血病、骨髓增生异常综合征、紫癜、过敏性紫癜、风湿、免疫功能失调、铁缺乏、急性鼻咽炎、病毒感染、溶血、细菌感染、白细胞减少症、原发性血小板增多症、脾脏肿大、营养不良、白细胞增多、原发性血小板减少症、肝炎、结缔组织病、颈部淋巴结结核
 可咨询
可咨询
擅长:贫血、再生障碍性贫血、缺铁性贫血、溶血性贫血、地中海贫血、营养性贫血、小儿贫血、紫癜、血小板疾病、血小板减少症、血小板增多症、免疫性血小板减少症、血液系统肿瘤、白血病、慢性粒细胞性白血病、骨髓增生异常综合征、慢性白血病、急性淋巴细胞性白血病、骨髓增生性疾病、红细胞增多症、骨髓纤维化、铁缺乏、溶血、儿童白血病、色素性紫癜、白细胞减少症、凝血障碍


日前,日本东京医科齿科大学宣布,利用老鼠的胚胎干细胞成功培养出了迷你心脏。研究小组称,在培养过程中,胚胎干细胞分化出了心肌细胞,并在大约两星期里发育成了直径约1毫米左右的心脏类器官。它和老鼠胎儿的心脏类似,具备心房及心室等构造,能像真正的心脏那样有规律地跳动,令人惊奇。
2020-09-15 生活

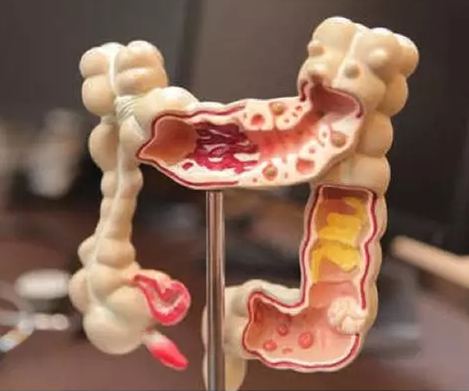
息肉是一个肉眼看到的诊断名词,没确定具体性质前都可以用这个名称,但这样的一个突起,在显微镜下,息肉可以是炎症,也可以是肿瘤。在未确定其病理性质之前统称为息肉,明确病理性质后则按部位直接冠以病理诊断学名称,如结肠管状腺瘤、直肠原位癌、结肠炎性息肉等。
Copyright © 2011-2026 北京春雨天下软件有限公司 All Rights Reserved
京ICP证120150号 京ICP备12050281号-1
(京)网药械信息备字(2023)第 00530 号
违法和不良信息举报:4006865590转9 feedback@chunyu.me






